2019年全球糖尿病患病率约为9.3%(4.63亿人),预计到2030年将上升到10.2%(5.78亿),到2045年将上升到10.9%(7亿)。糖尿病(DM)患者发生心力衰竭的风险很高,即使在校正了冠状动脉疾病或高血压之后也是如此。这导致了人们对一种独特的疾病过程的认识增加,这种疾病被定义为糖尿病心肌病(DCMP)。
DCMP的发生和发展是复杂的,涉及早期舒张期功能障碍、心肌肥厚、心室扩张和收缩功能障碍。临床上,以舒张期功能障碍为特征的射血分数保留性心力衰竭(HFpEF)也可能是DCMP的早期表现,这加强了机制研究的重要性和有效干预的迫切需要。
2022年12月14日,华中科技大学附属同济医院心内科的研究者们在Molecular Therapy: Nucleic Acids杂志上发表了题为“Positive feedback loop of miR-320 and CD36 regulates the hyperglycemic memory-induced diabetic diastolic cardiac dysfunction”的文章。该研究使用了集萃药康斑点鼠(BKS-Leprem2Cd479/Gpt),发现MiR-320和CD36以正反馈环的形式调节高血糖记忆诱导的糖尿病舒张性心功能障碍。

研究发现一:使用胰岛素控制血糖不能挽救链唑霉素所致的糖尿病舒张性心功能障碍
研究者使用链唑霉素(STZ)诱导增加C57BL / 6J小鼠的循环葡萄糖水平,分别在诱导4周、8周、12周评估心脏性能,发现诱导8周心脏有明显的舒张功能障碍;于是研究者在8周安排胰岛素注射,并连续注射4周,发现胰岛素注射并未能使心脏舒张功能恢复正常。并且研究者在STZ诱导4周(心脏还没有表现出舒张功能障碍的)时,安排胰岛素注射4周后发现心脏舒张功能障碍仍然会出现。这些发现表明,在高血糖应激4周后,胰岛素治疗不再能够预防或逆转舒张功能障碍,表明高血糖应激4周后已经建立了高血糖记忆。(图1)

图1.胰岛素控制血糖不能挽救STZ所致的糖尿病舒张性心功能障碍
研究发现二:miR-320下调可防止STZ诱导的心脏舒张功能障碍
研究者通过在STZ诱导的糖尿病小鼠心脏中对高血糖相关miRNA的表达测定发现miR-320是最丰富的,且表达不能通过胰岛素治疗恢复正常。于是研究者使用重组腺相关病毒系统对小鼠心脏递送miR-320抑制剂(TUD)来达到抑制miR-320表达的目的,发现miR-320表达抑制后,小鼠显示出心脏舒张功能改善。(图2)
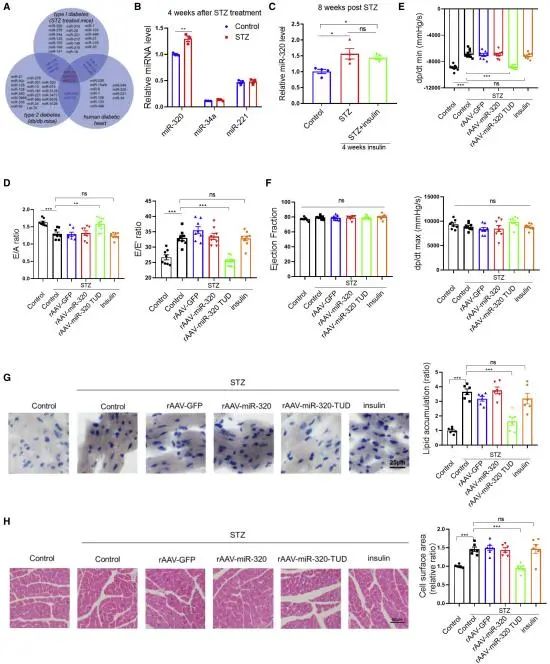
图2.miR-320的下调可防止STZ诱导的心脏舒张功能障碍
研究发现三:miR-320下调可防止db / db小鼠的心脏舒张功能障碍
研究者使用了db / db小鼠(一种成熟的2型糖尿病(T2DM)小鼠模型)来进一步研究miR-320在高血糖记忆中的作用,与STZ诱导的1型糖尿病(T1DM)类似,胰岛素治疗降低了db / db小鼠的血糖水平,但未能逆转miR-320水平和舒张功能障碍。相比之下,miR-320下调显著改善了db / db小鼠的心脏舒张功能。(图3)

图3.miR-320下调可防止db / db小鼠的心脏舒张功能障碍
研究发现四:心肌细胞CD36/miR-320正反馈回路
CD36是miR -320介导的细胞核转录激活的关键下游靶基因,本研究中CD36过表达增加了培养心肌细胞中miR-320的表达,而CD36敲低则降低了miR-320的水平。为了阐明CD36介导的miR-320激活的具体分子机制,研究者使用JASPAR和TRANSFAC 2.0数据库筛选了可以直接增强miR-320转录的潜在转录因子(SP1、GATA4、C-MYC、SMAD和RXR),并对这些转录因子的表达量进行检测,发现高糖处理后心肌细胞中只有SP1水平显著升高,且SP1的表达增加了心肌细胞中miR-320的表达,此外CD36过表达也增加了SP1的表达并且SP1敲低后CD36则无法诱导miR-320的表达,这表明CD36对miR-320的调控依赖于SP1。
接下来研究者想要研究CD36增加心肌细胞SP1表达的机制,之前的报道发现CD36在巨噬细胞中加速ROS生成,而ROS在管状上皮细胞中通过PKC/MAKP途径激活SP1,于是研究者在心肌细胞AC-16中转染ROS清除剂NAC,发现CD36的过表达不能增加SP1的表达量。研究者在体内观察到STZ诱导小鼠8周后心脏miR-320和CD36蛋白水平升高。此外,使用胰岛素控制血糖4周后,这种增加并没有消除。这些数据表明高血糖心肌细胞中存在CD36/miR-320正反馈回路。(图4)
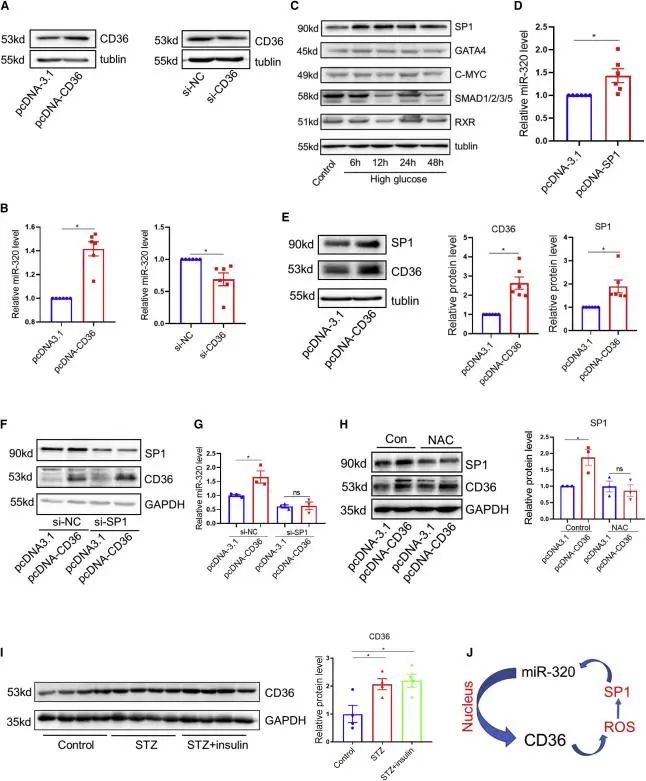
图4.心肌细胞CD36/miR-320正反馈回路
研究发现五:CD36 诱导在高血糖早期启动 CD36/miR-320 反馈回路
为了进一步确定CD36/miR-320反馈回路的触发因素,研究者通过对STZ处理小鼠心脏中CD36和miR-320表达的时间过程分析发现CD36上调(2周)先于miR-320(4周)。此外,高血糖诱导2周后,CD36 mRNA水平没有增加,这表明STZ给药2周后CD36蛋白的上调是通过转录后调控实现的;体外实验中,高糖处理AC-16细胞最早在6小时内迅速诱导CD36蛋白表达,但直到48小时后才影响CD36 mRNA表达。随后研究者通过多聚核糖体分析进一步证明了葡萄糖应激在早期阶段增强了CD36的翻译。(图5)

图5.CD36诱导在高血糖早期启动CD36/miR-320 反馈回路
研究发现六:Ago2缺失介导高血糖对CD36的早期诱导
为了研究CD36在高血糖早期的翻译上调的调控机制,研究者测试了Ago2(一种核心RNA诱导沉默复合物(RISC)成分,通常在翻译水平上负调节基因表达)是否参与了这一过程,观察到高糖处理3小时后心肌细胞Ago2表达显著降低。体内研究显示早期糖尿病患者心脏Ago2水平下降。且心肌细胞Ago2敲除可增加CD36蛋白水平,但不影响其mRNA表达。RNA免疫沉淀(RIP)显示高糖处理心肌细胞中CD36 mRNA与Ago2的结合减少,表明高血糖通过抑制Ago2的表达,导致CD36 mRNA与Ago2/RISC分离,从而激活CD36的翻译。
接下来研究者又进一步探索了高血糖导致Ago2在心肌细胞中缺失的机制,首先研究者发现高血糖不影响心肌细胞中Ago2 mRNA的水平,且早期翻译活性也保持不变,于是研究者检测细胞培养上清中Ago2蛋白水平,发现高血糖处理3小时后,细胞培养上清液中Ago2蛋白水平增加;此外研究者还发现高糖诱导后泛素化Ago2蛋白水平增加,且使用泛素化抑制剂MG132处理AC-16心肌细胞可有效阻止高糖导致的Ago2下调。
这些数据表明,在糖尿病早期,CD36的翻译上调可能是通过Ago2的减少来实现的,Ago2的减少可能是由于向细胞外空间的分泌增加和泛素-蛋白酶途径介导的蛋白降解增强。(图6)

图6.Ago2缺失介导高血糖对CD36的早期诱导

研究表明,CD36和miR-320之间存在正调节环路,CD36诱导是后来诱导miR-320表达的早期触发因素。此外,DCMP早期CD36增加可能是由参与靶向降解的Ago2蛋白减少介导的。
在糖尿病心肌病(DCMP)中,miR-320和CD36相互增强彼此的表达,导致心肌细胞中出现正反馈回路和可能的高血糖记忆。miR-320抑制可以挽救糖尿病小鼠的舒张功能障碍。这些发现为通过靶向miR-320治疗DM诱导的心功能不全的潜在治疗策略提供了概念验证证据。

 官方订阅号
微信公众号
官方订阅号
微信公众号